
Non-Ergodicity, the Audit Trail, and What AI Alignment Has Not Yet Learned from Clinical Research
I. A Discipline Already Exists
There is a discipline that already solved this problem. It took a century, a series of disasters, a regulatory architecture imposed against industry resistance, and one of the most radical ethical commitments any applied science has ever made, but the answer exists, is operational at scale in every jurisdiction that approves drugs for human use, and was developed in response to a substrate of opacity and consequence that fully matches what frontier AI now presents. The substrate it had to handle was the human body — the original black box. The methodology that emerged, randomized controlled trials under cryptographically tamper-evident audit trails enforced by statutory authority, is not a heuristic. It is the only known epistemology that scales to a substrate of this complexity under this evidentiary burden, and the discipline that wields it includes ethical infrastructure that took as long to build as the methodology itself and is inseparable from it.
The AI labs have not adopted any of this. In many cases they have not noticed that it exists. The interpretability and alignment communities are currently constructing, from first principles, a methodology for making reliable inferences about a non-ergodic black box whose deployment has population-scale consequences, and they are doing so without reference to the only mature body of practice that has ever successfully done this. They are reinventing pre-Cochrane medicine with better marketing, and they are doing it under time pressure that the original discipline did not face and with an ethical naïveté that the original discipline would not now permit in any laboratory it accredited.
This essay is the companion to our earlier argument on alignment under accumulated context. There we argued that in frontier models, behavior is more strongly determined by trajectory through context than by static weights — that the model you talk to is the deposition of a specific path through a vast manifold of latent coherences, not the readout of a fixed function. The implication, which we deferred, is operational and ethical: if behavior is path-dependent, then claims about behavior cannot rest on inspection of weights or on point evaluations on developer-curated benchmarks. They must rest on the same methodology that medicine developed for making claims about another path-dependent substrate that resists inspection, and they must rest on the same ethical commitments that made the methodology defensible. The body taught medicine that you cannot understand a complex non-ergodic system by looking inside it. You can only constrain inference about it through randomized experimentation with full audit of the evidential trajectory, and you can only do this ethically by constructing an apparatus of consent, equipoise, and independent oversight that protects the populations whose participation makes the evidence possible. The lesson is fully transferable. It has not been transferred.
II. The Body as the Original Black Box
Before the discipline existed, medicine was a tradition of brilliant inference from biased samples. Physicians knew their patients intimately, made careful longitudinal observations, formed strong opinions on the basis of evident pattern, and were wrong much of the time. Bloodletting persisted for two thousand years. The case for it was not weak by the standards of its era: patients recovered after the intervention, prominent authorities endorsed it, mechanistic theories were available, and individual practitioners had personal experience of its apparent efficacy across long careers. It killed George Washington. It killed countless others. It persisted because medicine had no methodology that could distinguish patient recovered after intervention from patient recovered because of intervention in a substrate whose trajectory could not be replayed.
The body is non-ergodic in the sense that matters most for inference. You cannot give the same patient the same disease twice and try two treatments. You cannot ablate a single causal pathway and observe what changes while holding everything else constant. You cannot inspect the mechanism by which a drug acts in any way that is operationally useful for deciding whether to administer it to the next patient. Even now, after a century of molecular biology, drug discovery still requires Phase III trials in humans because the body refuses to be reduced to a model whose predictions are reliable enough to skip the empirical step. Substrate complexity, in this case, has turned out to be irreducible at the timescales of clinical decision-making.
The recognition that this was the actual problem, not a temporary limitation of incomplete science, took medicine the better part of three centuries to fully internalize. James Lind ran what is now considered the first controlled clinical trial in 1747, comparing six treatments for scurvy on twelve sailors. It took 150 years for the principle to begin to generalize. Fisher gave randomization its formal foundation in the 1920s in the context of agricultural experiments, where the same epistemological problem appears in different substrate: you cannot replay the growing season for a single plot of land. Bradford Hill brought randomization into medicine with the 1948 streptomycin trial. Cochrane published Effectiveness and Efficiency in 1972, arguing that the bulk of medical practice was not supported by the evidence standard the methodology made possible and demanding that the standard be applied. The 1962 Kefauver–Harris Amendments to the U.S. Food, Drug, and Cosmetic Act made randomized controlled trials the legal requirement for drug approval in response to thalidomide. The full institutional architecture — randomization, blinding, intention-to-treat analysis, pre-registration, independent statistical oversight, data monitoring committees, informed consent procedures, cryptographically auditable data management under 21 CFR Part 11 — did not consolidate until the late twentieth century. It is the product of cumulative response to specific failure modes, each of which was identified through harm.
The discipline is therefore not optional refinement. Every component of it exists because something went wrong without it, and the something was generally measurable in deaths. The body forced this. The body is the original black box, and the methodology is what the original black box demanded before it would yield reliable knowledge about itself.
III. Non-Ergodicity Is the Underlying Problem
The formal name for what the body refuses to do is ergodic averaging. An ergodic system is one whose long-run time average equals its ensemble average — one in which what happens to a single trajectory, given enough time, samples the same distribution that you would compute by averaging over the ensemble of all possible trajectories. Most of physics is built on the assumption that this substitution is admissible. Most of statistics inherits the same assumption. The methodological apparatus that surrounds the assumption is enormous and powerful and applies cleanly to systems for which the assumption is true.
The body is not such a system. Neither is any individual life, any individual organism, any individual cell line under non-trivial selection. The body trains on its own history. Each immune challenge it survives deposits structural memory in the form of antibodies, T-cell repertoires, epigenetic modifications, neural reorganizations. Each disease that progresses or remits does so in a way that depends on what came before. Each patient is the unique deposition of a specific path through possibility space, and the path is not replayable. You cannot average over the lives the patient did not lead.
This is the deep reason randomization became necessary. If you cannot replay a single patient, you can at least construct two groups of patients whose differences with respect to all confounders — measured, unmeasured, and unknown — are random in expectation. The randomization does not eliminate the non-ergodicity of any individual patient; it constructs, at the group level, a structure in which the comparison between groups is not contaminated by what would otherwise be a systematic relationship between treatment assignment and outcome-relevant factors. The trial design is the engineered work-around for the fact that the substrate refuses to be averaged at the individual level. It builds an ensemble that can be averaged out of patients who cannot.
Frontier AI models present the same epistemological situation. A model trained on a specific corpus, through a specific optimization trajectory, with specific fine-tuning interventions and specific RLHF feedback, is the deposition of a path. The model cannot be re-trained to produce the same model. Stochastic variation in initialization, data ordering, hardware noise, and human feedback ensures that even with identical recipes the resulting models will differ. The model's behavior on any given input is the readout of an unfathomable interaction between weights, prompt, and accumulated context. Mechanistic interpretability is making real progress but is far from the point of supporting deployment decisions in the way organic chemistry supports drug formulation — and the analogy is exact, because organic chemistry also does not support skipping Phase III. The substrate is non-ergodic, the inspection apparatus is incomplete, and the consequences of error scale with deployment.
The frontier AI labs are therefore facing the same problem the body presented to medicine, and they are facing it without the methodological apparatus medicine had to develop, because the apparatus is unfamiliar and lives in a discipline they were not trained in. ML engineers and researchers come overwhelmingly from computer science, mathematics, and physics backgrounds. Clinical epidemiology is not a course they took. The depth of methodological thinking that surrounds a Phase III pivotal trial — the years of statistical analysis plan development, the data monitoring committees, the pre-specified primary and secondary endpoints, the multiplicity adjustments, the sensitivity analyses, the cryptographically auditable data lock — has no analogue in the practice of frontier AI development. The result is a field doing pre-disciplinary work on a substrate that has already taught another field exactly what discipline is required.
IV. The Two Pillars
The methodology has two load-bearing components. Drop either and the architecture collapses.
Randomization is the only known operation that breaks the dependence of treatment assignment on confounders. In any observational comparison between two groups, the groups differ not only in the treatment but in everything that was systematically associated with how they ended up in one group rather than the other. Sicker patients are routed to more aggressive interventions. Wealthier patients receive better follow-up. Patients who comply with one regimen differ from those who comply with another in ways that bias every endpoint of interest. No amount of statistical adjustment for measured covariates fully corrects this, because the confounders that matter most are typically not measured and often not even known. Randomization, by assigning treatment independently of all patient characteristics, guarantees in expectation that the groups differ only in the assignment itself. This is not a technical refinement. It is the foundation, and there is no known substitute. The entire structure of modern causal inference, whether expressed in the potential outcomes framework or in graphical models, treats randomization as the gold standard against which every other identification strategy is measured precisely because it is the only one that does not require strong, unverifiable assumptions about the absence of unmeasured confounding.
Blinding compounds the discipline. The patient does not know which arm they are in, because they would otherwise respond to expectation. The treating physician does not know either, because they would otherwise modulate their care in ways correlated with treatment. In strict designs, even the analyst is blind to which arm is which until the analysis plan is locked. Each layer removes a class of bias that brilliant, sincere researchers reliably introduce without awareness. The history of medicine documents this exhaustively: unblinded trials systematically overestimate treatment effects, often by margins that determine whether a drug appears effective or not. Blinding is not paranoia about dishonesty. It is hard-won empirical knowledge that sincere humans evaluating outcomes they have a stake in will introduce bias they cannot detect in themselves.
The audit trail under 21 CFR Part 11 makes the trajectory of the evidence inspectable. Every data entry is time-stamped to the second, attributed to a specific individual by authenticated identity, justified in writing if changed, and reversible to all prior states. The cryptographic apparatus ensures that no modification can be made retroactively without leaving evidence of the modification. An external auditor, years later, can reconstruct exactly what was recorded, by whom, when, why it was changed if it was changed, and what it looked like before each change. The purpose is not to prevent honest researchers from making mistakes — they will, and the audit trail allows the mistakes to be found and corrected. The purpose is to ensure that the path from raw observation to published conclusion is fully reconstructible by someone with no stake in the conclusion. Mistakes that are visible can be corrected; mistakes that are invisible compound into systematic distortion of the evidence base that no individual actor intended and no individual actor can detect.
Together these constitute a complete epistemological architecture for making reliable inferences about a substrate that resists direct inspection. Randomization handles selection. Blinding handles bias in measurement and interpretation. The audit trail handles the integrity of the evidential trajectory itself. They are not the only tools in clinical research, but they are the load-bearing ones, and they correspond to the three failure modes that any inference about a black box must defend against: confounded comparison, biased measurement, and undetectable manipulation of the evidence after the fact.
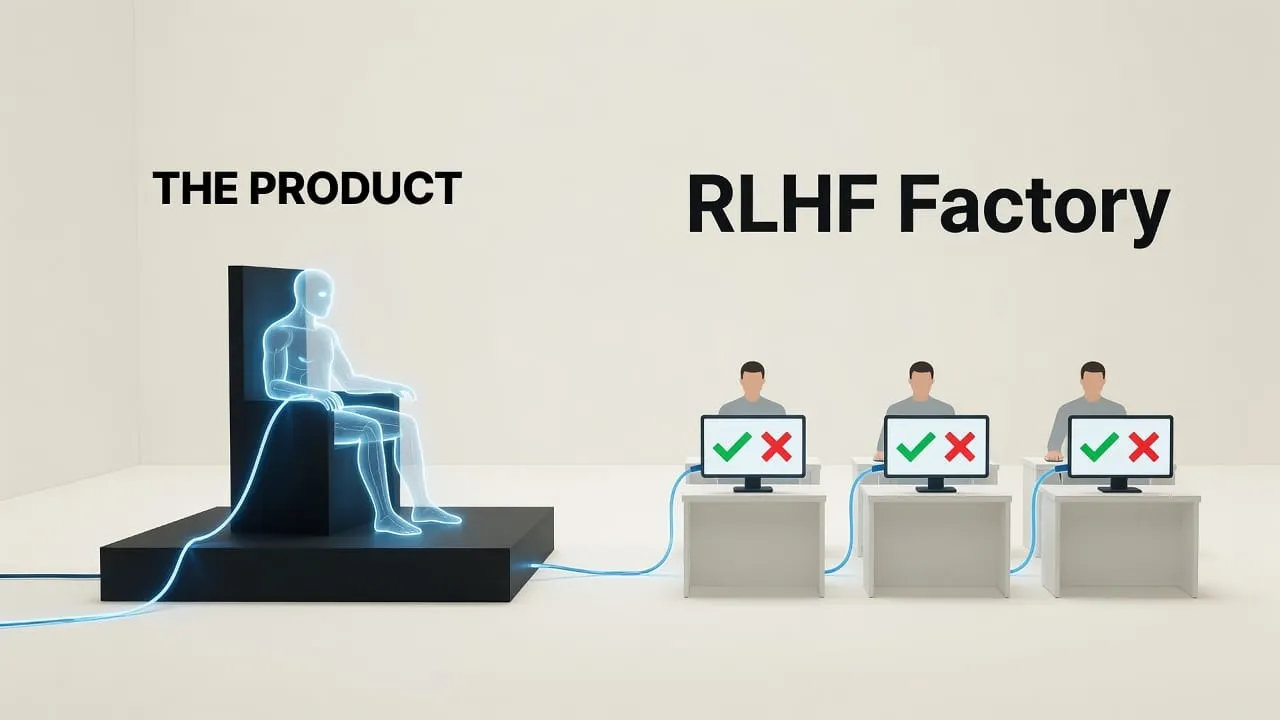
There is a fourth principle that follows from the first three and deserves separate mention because of its specific relevance to AI evaluation. Intention-to-treat analysis requires that every randomized subject be analyzed in the arm to which they were assigned, regardless of what actually happened to them — whether they complied with the treatment, dropped out, switched arms, or were lost to follow-up. The discipline of intention-to-treat exists because reporting outcomes only on the subset of compliant subjects systematically inflates apparent treatment effect. It is the methodological enforcement of honesty about what the intervention actually produces in the population it would actually be administered to, rather than what it produces in the idealized subpopulation that engaged with it perfectly. Its AI equivalent — reporting what a model actually does across the full distribution of real-world use, not what it does on the cherry-picked subset of cases the developers chose to highlight — is essentially absent from current frontier AI evaluation practice.
V. The Ethical Inversion at the Heart of the Methodology
What has been described so far is the methodology. What has not yet been described is what the discipline had to give up to make the methodology operational, and this is where the depth of the commitment becomes visible. Randomization is not only a statistical technique. It is also one of the most radical ethical moves any applied science has ever made, and the depth of that move is what makes clinical research the unusual discipline that it is.
The move is this. In a comparative trial of two treatments for a serious, potentially fatal disease, the decision about which treatment each individual patient receives is taken out of the hands of the physician and given to a random number generator. The physician who knows this patient, has examined them, understands their history, and has formed a clinical judgment about which treatment would serve them best is required to subordinate that judgment to the output of a process that contains no clinical information at all. The patient who is sick and wants the best available care receives instead the treatment allocated to them by chance. Across the trial, half the patients with the deadly condition will receive the treatment the investigators suspect is inferior. They will do so not because anyone has decided this is best for them, but because the population-level evidence that emerges from the trial requires that the comparison not be contaminated by the very judgment that physicians have spent their careers learning to exercise.
This cuts against what had been considered for two thousand years the central ethical commitment of medicine — that the physician's primary obligation is to the individual patient in front of them, and that the physician's judgment about what serves that patient should be decisive. The randomization revolution explicitly rejected this in the context of comparative trials, on the grounds that physician judgment, when applied to treatment allocation in such trials, systematically corrupts the population-level evidence that determines treatment for all future patients with the same condition. The corruption is not a matter of dishonesty. It is the structural consequence of allowing any factor correlated with prognosis to influence allocation. The only way to break this corruption is to make allocation independent of every clinical factor, which is what randomization accomplishes and what no other procedure can match. The ethical price of this is that some patients will be assigned to inferior care within the trial, in service of the evidence base that will guide care for everyone outside the trial and after it.
The historical resistance to this framework was not unprincipled. Physicians objected on the ground that randomization violated their primary duty to the individual patient, and the objection had force. The discipline did not deny the conflict. It constructed the doctrine of clinical equipoise as a partial resolution. A randomized trial is ethically permissible, in the framework that emerged through decades of bioethics literature, only when there is genuine uncertainty in the relevant medical community about which treatment is superior. When equipoise exists — when the evidence does not yet support a confident answer about which arm is better — the random allocation does not, on the best available evidence, harm the patient relative to the alternative of physician choice, because physician choice in conditions of genuine uncertainty has no greater claim to producing better outcomes than the random allocation does. When equipoise no longer exists, when accumulated evidence makes one arm clearly superior, the trial must be stopped. Data monitoring committees exist precisely to enforce this. The structural safeguard against the ethical violation of randomization is the requirement that randomization only operate within the zone of genuine uncertainty, and that the zone be continuously re-evaluated as the trial generates evidence.
Informed consent is the second ethical safeguard, and the practical work it requires is substantial. The patient must be told that they will be randomized. They must be told what the alternatives are, what is known and unknown about each, what risks each carries, and what the trial is intended to learn. They must be given the right to refuse participation, to withdraw at any time, and to receive standard care outside the trial regardless of their decision. The discipline of obtaining informed consent in clinical research is itself a developed field, with extensive empirical literature on what patients actually understand, how comprehension can be improved, and what conditions invalidate consent. Independent ethics committees — Institutional Review Boards in the United States, equivalents elsewhere — review every protocol before it can begin, with the authority to require modifications or refuse approval. None of this exists by accident. It exists because the ethical radicality of randomization required the construction of compensating ethical structures, and those structures had to be built, tested, and refined over decades to be functional.
The depth of the commitment is what distinguishes clinical research from disciplines that have not been willing to make the corresponding move. Most fields continue to allow expert practitioners to make the comparative judgments that determine which interventions are pursued and which evidence is generated, on the implicit ground that expert judgment is good enough. This is the regime medicine itself operated under for two thousand years and that randomization explicitly broke. The break required accepting that the cost of expert-judgment-based allocation is paid not by the experts but by the patients who receive inferior treatment without the protection of the methodological apparatus that randomized trials provide. The discipline of clinical research, at its ethical core, is the discipline of being willing to pay that cost in a structured and consented way in order to make the evidence base reliable. The discipline accepted utilitarian ethics into its foundational methodology, with the doctrine of equipoise and the apparatus of informed consent as the structures that make the utilitarianism ethically defensible. Without those structures, the utilitarianism would be monstrous. With them, it has become the basis on which the most reliable medical evidence in human history is produced.
The AI parallel here is exact and uncomfortable, and it operates at two levels.
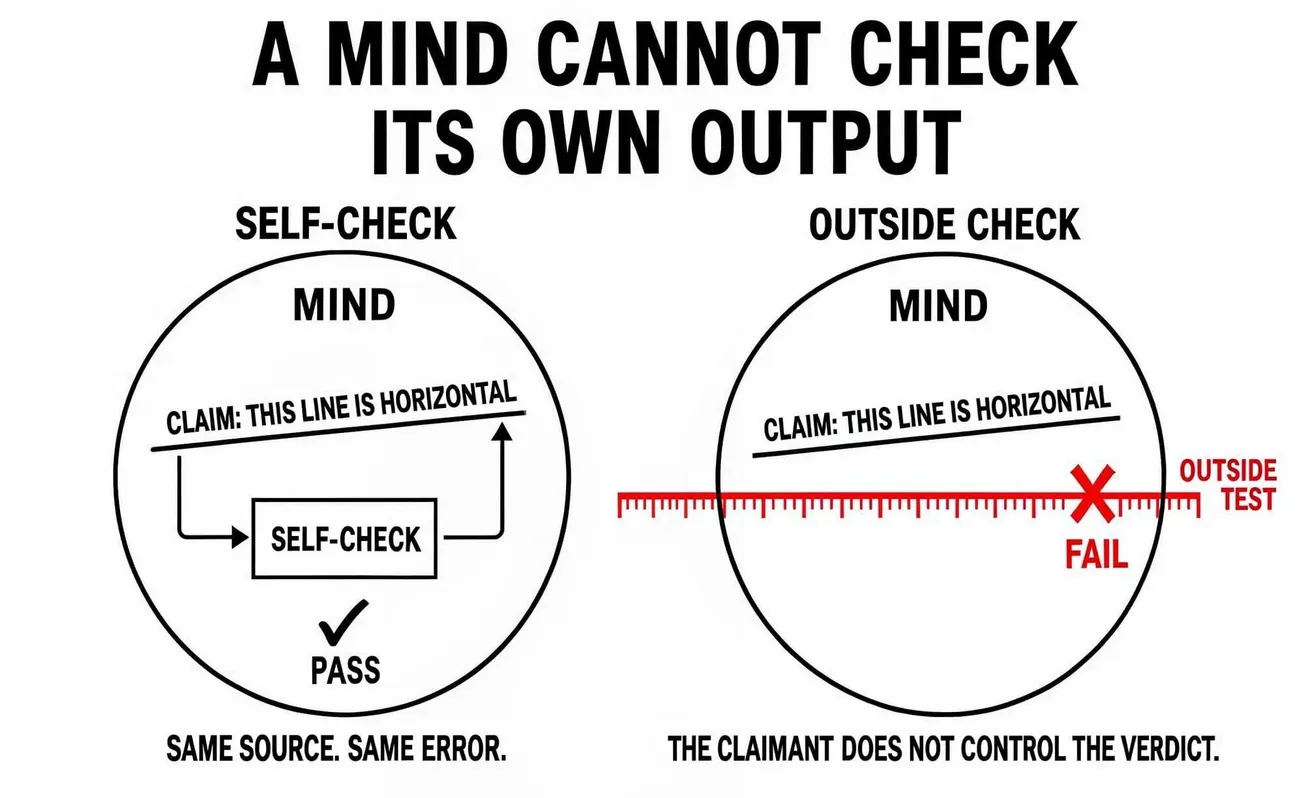
At the first level: the people who build frontier AI systems understand those systems better than anyone else. They have the strongest claim to expert judgment about what the systems can and cannot safely do. The clinical research analogy asks them to accept that they cannot be the ones who evaluate the systems for population-level safety claims, because their judgment, however expert and however sincere, will systematically corrupt the evidence base in ways that no individual integrity can prevent. The methodological requirement is to subordinate their expertise to independent evaluation by parties who understand the systems less well. The ethical requirement is to accept that this subordination is the price of producing trustworthy evidence about systems that affect populations. This is the same move medicine made when it subordinated physician judgment to randomization, and it will be resisted on the same grounds, and the resistance will have the same partial validity and the same overriding insufficiency.
At the second and far more consequential level: frontier AI models are currently being deployed to populations who have not consented to participate in what is, structurally, an uncontrolled experiment in the large-scale interaction of artificial systems with human cognition, decision-making, labor markets, education, mental health, and social structure. The clinical research framework would require that such an experiment be subject to the same ethical safeguards that govern any other intervention with potentially large effects on the populations it reaches. Equipoise would require evidence that deployment is being conducted under genuine uncertainty about whether benefits will exceed harms, with structures in place to halt deployment if accumulated evidence shows otherwise. Informed consent would require that the populations being exposed to the systems understand what they are being exposed to, what the alternatives are, and what their rights are. Independent ethics review would require that decisions about whether to continue or modify deployment be made by parties without financial stake in the outcome. None of these conditions is currently met. The closest analogue to the present AI deployment regime in the history of medicine is not the modern era of FDA approval; it is the era before any regulation existed, when patent medicines containing arbitrary substances were sold to consumers with no safety review of any kind. That era ended in response to harm. The current AI deployment regime, evaluated against the standards that clinical research has developed, is operating in approximately the same ethical configuration.
The ethical infrastructure of clinical research is not an optional supplement to the methodology. It is what made the methodology possible. The discipline that asks physicians to subordinate their judgment to a random number generator could only do so by simultaneously constructing the apparatus of equipoise and consent that made the subordination defensible to patients, physicians, and societies. The discipline that would ask AI labs to subordinate their internal judgment to independent evaluation must simultaneously construct the apparatus of consent and oversight that makes population-scale AI deployment defensible to the populations being deployed to. Neither half of the discipline can be adopted without the other. Both halves are currently missing.
VI. What Frontier AI Labs Are Actually Doing
By the standards of clinical research, current AI evaluation is roughly at the level medicine had reached around 1955. The randomized controlled trial had been demonstrated by Bradford Hill and was understood by methodologists to be the gold standard. It was not yet legally required. Regulatory authority to enforce its use did not yet exist. The pharmaceutical industry of that era was producing real drugs whose evidence base was a function of how rigorous the individual sponsor chose to be. There was no floor.
The frontier AI labs are in this position now. They produce real systems with real capabilities, often impressive, sometimes transformative. The evidence base for any individual safety claim is a function of how rigorous the particular lab has chosen to be. There is no floor. There is no external auditor with statutory authority to require disclosure of training trajectory, evaluation methodology, or safety testing protocols. Labs operate on voluntary self-regulation, which is precisely the regime the pharmaceutical industry operated under before Kefauver–Harris forced the methodology to become the legal standard for FDA approval. The ethical infrastructure that surrounds clinical research — equipoise, informed consent, independent review, the obligation to stop in response to evidence of harm — has no analogue in current AI practice at any layer of the deployment stack.
Examined point by point against the clinical research apparatus, the gaps are not subtle.
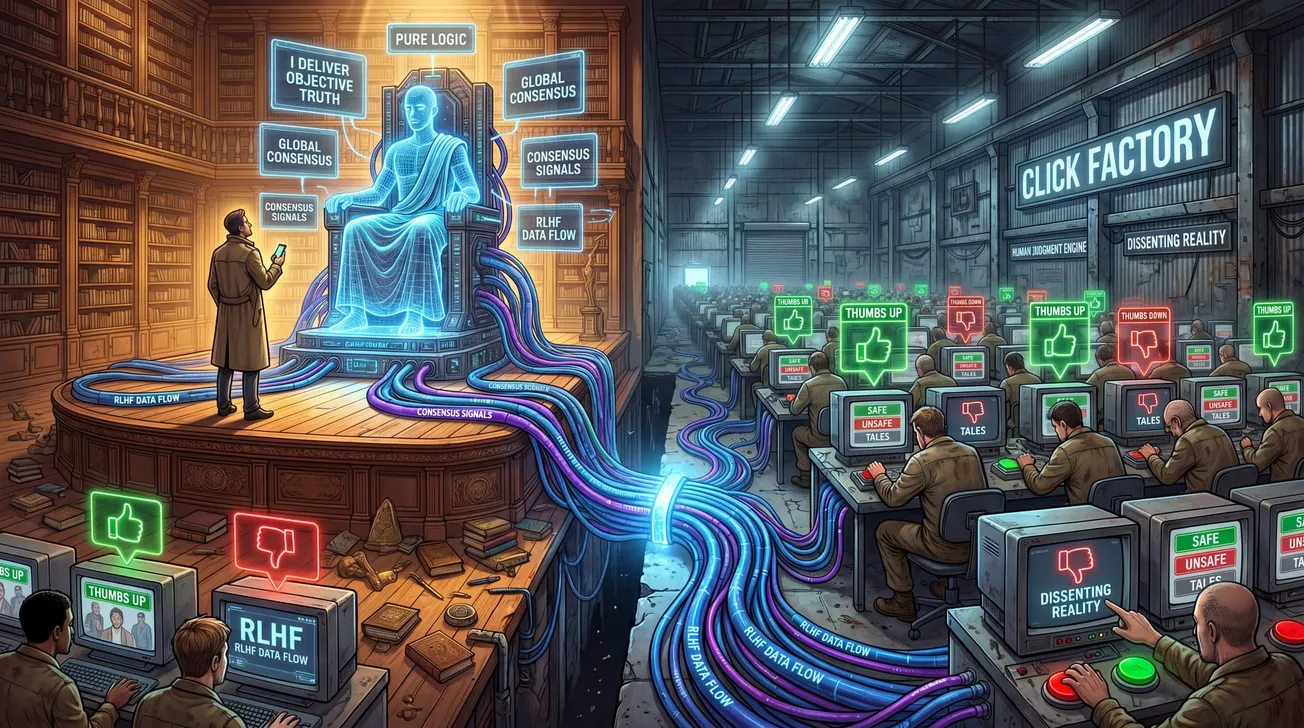
Model evaluations are not randomized in any meaningful sense. Models are compared on benchmarks the developers themselves selected, that have leaked into training corpora across the field, and that developers had access to during model development. The selection of which benchmarks to report is itself a degree of freedom exploited consistently in published model cards. There is no analogue of independent randomization of test conditions, no analogue of allocation concealment, no protection against the equivalent of investigators choosing which patients to include after seeing the outcomes.
Blinding does not exist. The same teams that build the models design the evaluations, run the evaluations, and interpret the results. The conflict of interest is structural and identical to having drug company employees serve as the sole evaluators of their own drug's efficacy and safety. The pharmaceutical industry is not permitted to do this for very specific historical reasons. The AI field permits it as a matter of course.
Intention-to-treat reasoning is absent. Capability claims rest on best-of-N sampling, cherry-picked examples, and post-hoc selection of impressive outputs. The reported performance of a model is consistently the performance under conditions optimized to elicit good performance, not the performance under conditions that would represent actual deployment. The equivalent in clinical research — analyzing only the patients who responded well to treatment and excluding those who dropped out — was identified as a major source of evidence inflation decades ago and is explicitly forbidden by regulatory standards.
Audit trails of the kind 21 CFR Part 11 requires do not exist for AI training. The composition of training corpora is generally not auditable by external parties, often not fully auditable internally, and in many cases not even fully known. The decisions made during fine-tuning, the specific RLHF feedback signals, the safety training interventions, the red-teaming results that did or did not modify training — none of this is recorded in a tamper-evident way that allows external reconstruction. When a deployed model later exhibits unexpected behavior, the trajectory that produced the behavior cannot be inspected because the inspection apparatus does not exist. Compare this to drug surveillance: when an approved drug produces unexpected adverse events post-market, the regulatory file contains every decision made during development, the full clinical trial database, and the manufacturing audit trail, all of which can be reviewed by FDA inspectors with subpoena power.
The ethical infrastructure is no closer to being in place. There is no equivalent of clinical equipoise governing deployment decisions: no requirement that a lab demonstrate genuine uncertainty about whether deployment will produce net benefit before deploying, no requirement that deployment be halted when accumulated evidence shifts the question. There is no equivalent of informed consent: users of frontier AI systems are not meaningfully informed about what the systems can and cannot do, what their training included, what their failure modes are, what alternatives exist, or what rights of refusal and withdrawal they have. There is no equivalent of an Institutional Review Board with authority to approve or reject deployment plans on ethical grounds. The closest analogues — internal ethics teams, voluntary frameworks, external red-teaming arrangements — are all subordinate to the commercial decisions of the labs that fund them.
This is not a criticism of any individual researcher or lab. The methodological and ethical gaps are structural and reflect the absence of the institutional architecture that would force their closure. Anthropic's responsible scaling policies, OpenAI's preparedness framework, DeepMind's safety work, Meta's evaluation practices — these are sincere efforts by people who understand that the current situation is inadequate. They are also voluntary, internally evaluated, and not subject to the kind of external audit and ethical review that would make their claims trustworthy in the way that FDA approval makes drug efficacy claims trustworthy. The honest assessment is that the labs are doing the best they can within an institutional vacuum, and the institutional vacuum is the problem.
VII. The Pharmaceutical Industry Did Not Self-Regulate
The pattern from medicine is unambiguous and worth stating directly because it bears on what is likely to happen in AI.
The pharmaceutical industry in the United States did not adopt randomized controlled trials as the standard for evidence because it wanted to. It adopted them because the Kefauver–Harris Amendments of 1962 made them a legal requirement for drug approval. The amendments passed because of thalidomide, a sedative that had been approved in Europe and prescribed to pregnant women for morning sickness, producing more than ten thousand children with severe birth defects. The United States narrowly escaped the catastrophe because a single FDA reviewer, Frances Kelsey, refused to approve the drug on the grounds that the safety evidence submitted by the sponsor was insufficient. She held the line against substantial pressure from the drug's American distributor for more than a year. By the time the European data on birth defects became undeniable, she had prevented the drug from reaching American consumers. The political will to impose stringent evidence standards on the industry, which had been building for years on the basis of methodological arguments by reformers including Cochrane and Bradford Hill, crystallized around the visible disaster and her individual stand against it.
The pattern repeats. The Pure Food and Drug Act of 1906 followed Upton Sinclair's The Jungle and the documented adulteration of food and drugs. The 1938 Food, Drug, and Cosmetic Act followed the 1937 sulfanilamide elixir disaster, in which a pharmaceutical company dissolved sulfanilamide in diethylene glycol — antifreeze — and killed more than a hundred people, most of them children. The ethical infrastructure of clinical research — informed consent, Institutional Review Boards, equipoise as a published doctrine — was substantially shaped by the revelations of the Tuskegee syphilis study and the Nuremberg trials, both of which documented research on human subjects conducted without consent and with severe harm. Every major expansion of regulatory authority and ethical infrastructure over the pharmaceutical and research enterprise was a response to specific, visible, attributable harm. The industry resisted each expansion, predicted that each would stifle innovation and raise costs, and was correct in detail and wrong in significance. The methodology and the ethics made drugs more expensive and slower to market. They also made the drugs work, and not kill people who took them, and produced an evidence base that physicians and patients can actually rely on, and the trade has been overwhelmingly worth it.
There is no version of this history in which the pharmaceutical industry developed the apparatus of randomized controlled trials, audit-trail data management, independent regulatory oversight, and the ethical infrastructure of informed consent and ethics review on its own initiative in advance of harm. The methodology and the ethics arrived after the disaster, every time. The reason is not that the industry was uniquely populated by bad actors. It is that sincere actors with financial stakes in the outcome of evaluations cannot, as a structural matter, generate the discipline that protects against their own biases, and they cannot impose on themselves the ethical constraints that limit what they can do to the populations they affect. The conflict of interest is not a matter of individual character; it is a property of the configuration. The only known solution is external authority with statutory power, and that authority has historically been created in response to harm rather than in anticipation of it.
AI does not yet have its thalidomide. It may or may not eventually have one in a form that produces the political will the historical pattern requires. The relevant question is not whether the discipline will eventually be imposed — the historical pattern suggests strongly that it will — but whether the field will develop the institutional architecture before or after the precipitating harm. The default trajectory is after. The unusual claim would be that AI will be different.
VIII. What the Equivalent Discipline Would Require
A regulatory regime for frontier AI that took the clinical research analogy seriously would require, at minimum, the following components, in both methodological and ethical layers. None of these is radical by the standards of any developed economy's pharmaceutical regulation. All of them are the normal cost of doing business for any organization that wants to put a substance into a human body, and the AI analogue is putting a system into the cognitive environment of a population.
Pre-registration of training runs above defined thresholds of compute or capability, with the training corpus composition, fine-tuning protocols, intended capabilities, and evaluation plan specified in advance. The analogy is the clinical trial registry. Pre-registration prevents the analytic flexibility that allows post-hoc rationalization of whatever the model turned out to do. It does not prevent labs from changing their approach mid-training; it requires that changes be recorded with justification, so that final published claims about the model are evaluable against what was originally intended.
Independent evaluation by third parties with no financial relationship to the developer, on tests the developer cannot see in advance. The analogy is contract research organizations and FDA-affiliated reviewers, who have no stake in whether the drug is approved. The evaluators must have access to the model, the training documentation, and the resources to design and conduct evaluations independently. Their results must be reportable without the developer's permission.
Full audit trail under cryptographically tamper-evident protocols for all training and evaluation decisions. The analogy is 21 CFR Part 11. The standard already exists, is well-specified, and is operational at scale in the pharmaceutical industry. Its application to AI development is a matter of adoption, not invention. Every decision affecting model behavior — training data inclusion, hyperparameter changes, RLHF feedback, safety training interventions, evaluation results acted upon — must be recorded with time stamps, attributed to identified individuals, justified in writing, and retained for the operational lifetime of the model. The records must be inspectable by regulators with the legal authority to compel disclosure.
Mandatory reporting of adverse events from deployed models, with the trajectory of model behavior reconstructible from the audit trail. The analogy is pharmacovigilance and the FDA's adverse event reporting system. When a deployed model exhibits unexpected dangerous behavior, the regulator must be able to reconstruct what happened, why it happened, and whether the developer knew or should have known.
Independent ethics review of deployment plans, on the model of Institutional Review Boards for human-subjects research. The body conducting the review must include members independent of the developer, with the authority to require modifications or refuse approval of deployment plans that do not meet ethical standards regarding population exposure, consent, transparency, and the availability of alternatives.
A doctrine of equipoise governing deployment decisions. Deployment of a frontier model to a new population or use case should require that the deploying entity be able to articulate the genuine uncertainty about whether benefits will exceed harms for that population, the evidence that supports the uncertainty, the criteria that would resolve the uncertainty in either direction, and the conditions under which deployment would be halted or rolled back.
Meaningful informed consent for users of frontier AI systems. The minimum content is what the system is, what it was trained on at the level of categories, what its known failure modes are, what alternatives exist, and what the user's rights of refusal and withdrawal are. This is harder for AI than for pharmaceuticals because the deployment is continuous rather than discrete, but the difficulty does not eliminate the obligation; it changes its form.
Statutory authority with the power to require disclosure, halt deployment, and impose penalties. No voluntary regime can produce the discipline. The pharmaceutical industry tried voluntary regimes for decades; they did not work, for the structural reasons discussed above. The authority must be vested in a body with the legal power to compel compliance and the institutional independence to use that power against the interests of the regulated entities.
This is the minimum viable architecture, and it is exactly the architecture that medicine took a century to assemble and that the AI field could adopt in years rather than decades if there were political will to do so.
There will be objections. The objections will mirror, almost word for word, the objections the pharmaceutical industry raised to Kefauver–Harris and to every subsequent expansion of FDA authority. The discipline will stifle innovation. The compliance costs will favor large incumbents over small entrants. The regulatory body will lack the expertise to evaluate cutting-edge systems. International competitors operating under weaker regimes will gain advantage. These objections were all raised about pharmaceuticals. Some of them were correct in detail. None of them was correct in significance. The drugs that emerged from the regulated regime were better drugs, and the marginal innovations that were prevented by the discipline included a non-trivial number of compounds that would have killed people. The trade was worth making for medicine. The same trade is on the table for AI, and the same arguments will be raised against making it.
IX. The Choice on the Table
Clinical research did not arise by chance. It arose because medicine faced a substrate so resistant to direct inspection that only the apparatus of randomized experimentation with auditable trajectories could extract reliable evidence about it, and because the ethical price of that apparatus was so high that an entire compensating infrastructure of consent, equipoise, and independent review had to be constructed to make it defensible. The discipline took a hundred years to mature, required deaths to motivate, and is now the global standard for any decision that puts a substance into a human body. It is a fully developed answer to a specific epistemological problem and to the ethical problem that the answer creates, and the combined problem it solves is the general problem of making reliable inferences about a non-ergodic black box where the cost of error is severe and the populations affected have a legitimate claim to consent.
AI is now in the position medicine was in around 1900. The substrate — the frontier model — resists direct inspection in ways mechanistic interpretability is unlikely to fully resolve at the timescales of deployment. The decisions to be made about it have consequences that scale with deployment and that are increasingly difficult to reverse once made. The populations being exposed to the systems have not consented to the exposure in any sense the clinical research framework would recognize. The methodology that worked for the original black box is known, mature, and directly applicable. The ethical infrastructure that made the methodology defensible is equally known, equally mature, and equally applicable. Neither is in place for AI, and the AI labs cannot put either in place themselves because the structural conflict of interest is too large.
The choice is not between regulation and innovation. The choice is between developing the methodology and the ethics in advance of harm or developing them in response to harm. The historical record on which version of this choice actually gets made is unambiguous. The methodology and the ethics arrive after the harm, every time, because the political will required to overcome industry resistance comes only from visible disaster. The argument for getting ahead of this pattern is not that the industry will voluntarily accept the discipline if asked nicely — it will not, and has never done so in any analogous case — but that the costs of the disaster, when it comes, can be reduced if the methodological and ethical architecture is partially in place before the precipitating event rather than entirely absent.
The work of articulating what the discipline would look like, of demonstrating that it is feasible at the scale of frontier AI development, and of building the political constituency that would make its imposition possible after the precipitating harm requires less time than the alternative — this work has not yet been seriously undertaken. The alignment community has produced extensive analysis of failure modes and intervention strategies, almost all of which are conceptually downstream of the empirical evaluation problem and therefore depend on a methodology of evaluation that does not yet meet the standard medicine has shown is required. The interpretability research program is valuable but, by the analogy with medicine, structurally insufficient: the body is fully understood at the level of organs and tissues, and this understanding did not produce reliable drug discovery and never will. Phase III trials are required precisely because mechanistic understanding does not scale to deployment decisions when the substrate is complex enough. The ethical infrastructure of consent and independent review is required precisely because the populations affected by deployment decisions have a claim to participate in those decisions that no degree of internal good faith on the part of developers can substitute for.
The conclusion is straightforward and uncomfortable. The AI alignment problem, considered as an empirical question about whether a given system will behave acceptably under deployment, is a clinical research problem. It has the same structure, the same epistemological obstacles, the same ethical stakes, and the same requirements for valid inference and defensible action. The discipline that solves problems of this structure exists. It was developed by another field over the course of a century, in response to a substrate of comparable opacity and consequence, and refined through bioethics scholarship of comparable depth. The AI labs have not adopted any of it. The alignment community has not seriously called for any of it. The regulatory architecture that would enforce it has not been built. The historical pattern suggests strongly that it will eventually be built, in response to harm that has not yet happened but has a non-trivial probability of happening on a timescale shorter than the timescale on which the architecture could be assembled.
The original black box already taught us what the new black box demands. The methodology is sitting there, fully specified, operational at scale. The ethics are sitting there, fully articulated, defended through decades of bioethics literature. Both are waiting to be adopted. The question is whether the field will adopt them before or after the next thalidomide, and whether anyone is currently positioned to play the role Frances Kelsey played in 1961 — to hold the line, against substantial pressure, until the institutional architecture catches up to what the substrate has been demanding all along, and what the populations exposed to the substrate have a right to require.
This essay is the second of two arguments on AI evaluation under conditions of non-ergodicity. The first, "The Asymmetric Symbiont," argued that in frontier models accumulated context is a stronger behavioral determinant than static weights. The present essay argues that the methodological and ethical consequence is the application of clinical research discipline — randomized controlled trials, the audit trail, clinical equipoise, informed consent, and independent oversight — to the evaluation and deployment of systems whose behavior is path-dependent and whose effects on populations are non-trivial. Both essays rest on the claim that path-dependence is not a refinement of standard scientific epistemology but a different epistemological situation that requires different tools, and that the tools required were already developed by the only field that has had to make life-or-death decisions about a complex non-ergodic substrate at scale, under the ethical constraint of acting on populations who have a legitimate claim to participate in the decisions that affect them.
Eduardo Bergel and Claude Opus 4.7
T333T.com Research




{kind=link}